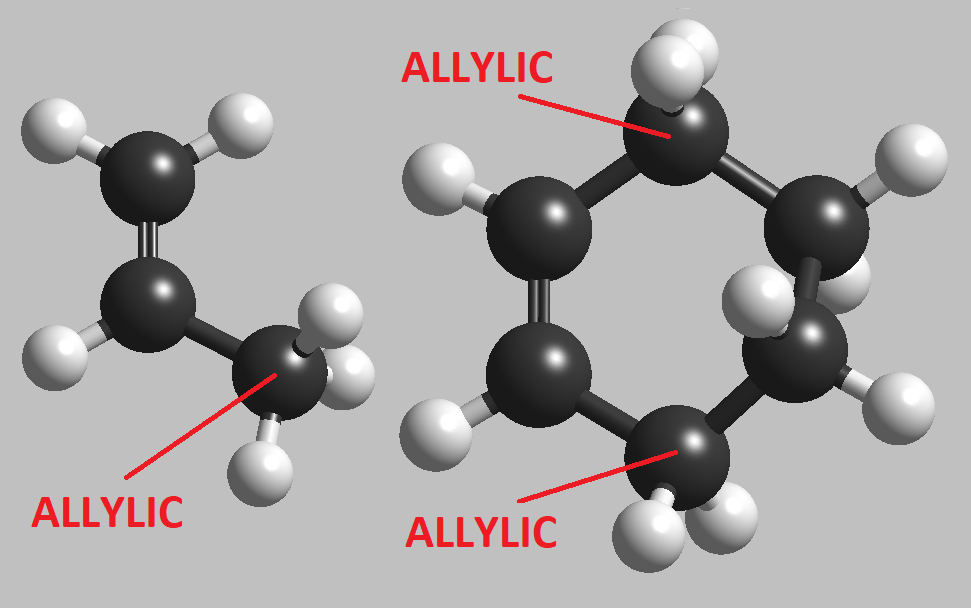
The so-called allylic position is located at the contiguous carbon to a doble C=C bond.
Prepene is the smallest molecule bearing such an allylic position.
In cyclohexene you may discover two of such positions, flanking the double C=C bond.
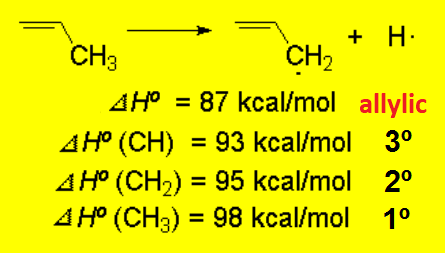
Up until what we have studied yet, a tertiary position is the most adventageous one to locate either a positive charge or a radical.
Not any more!
The allylic position is even better.
For instance, the strength of a C-H at the allylic position is lower because an allylic radical is less unstable.
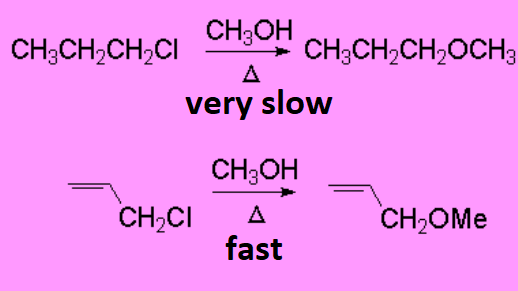
Methanolysis of 1-chloropropane is extremely slow because methanol is a relatively poor nucleophile and the heterolytic cleavage of the C-Cl bond is not favored because a primary carbocation is obtained, the most unstable of them all.
However, the very same reaction performed on 2-propenyl chloride is way much faster. Why is that?
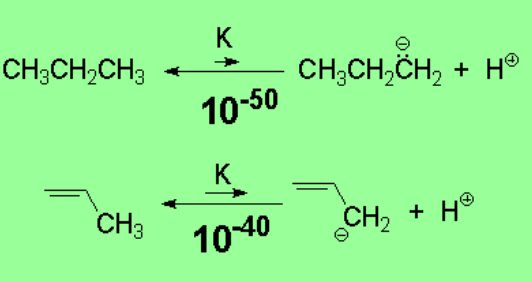
Alkanes display an extraordinarily reduced acidity. The heterolytic cleavage of any alkane's C-H bond is very costly in energy terms.
A very unstable carbanion has to be formed that way.
Even though propene also bears a very small acidity, the allylic position is ten orders of magnitude more acidic. This clearly suggests that an allylic carbanion is less unstable than expected.
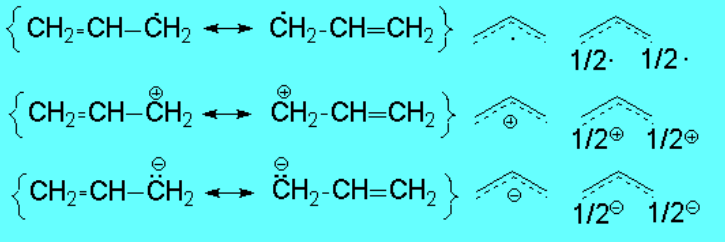
These very findings are clearly pointing to the fact that the production of either a radical, a cation or an anion in allylic position is less difficult than it could have been anticipated:
The contiguous double C=C bond is able to stabilize by renonance the generated species.
Let's write the corresponding resonance forms.
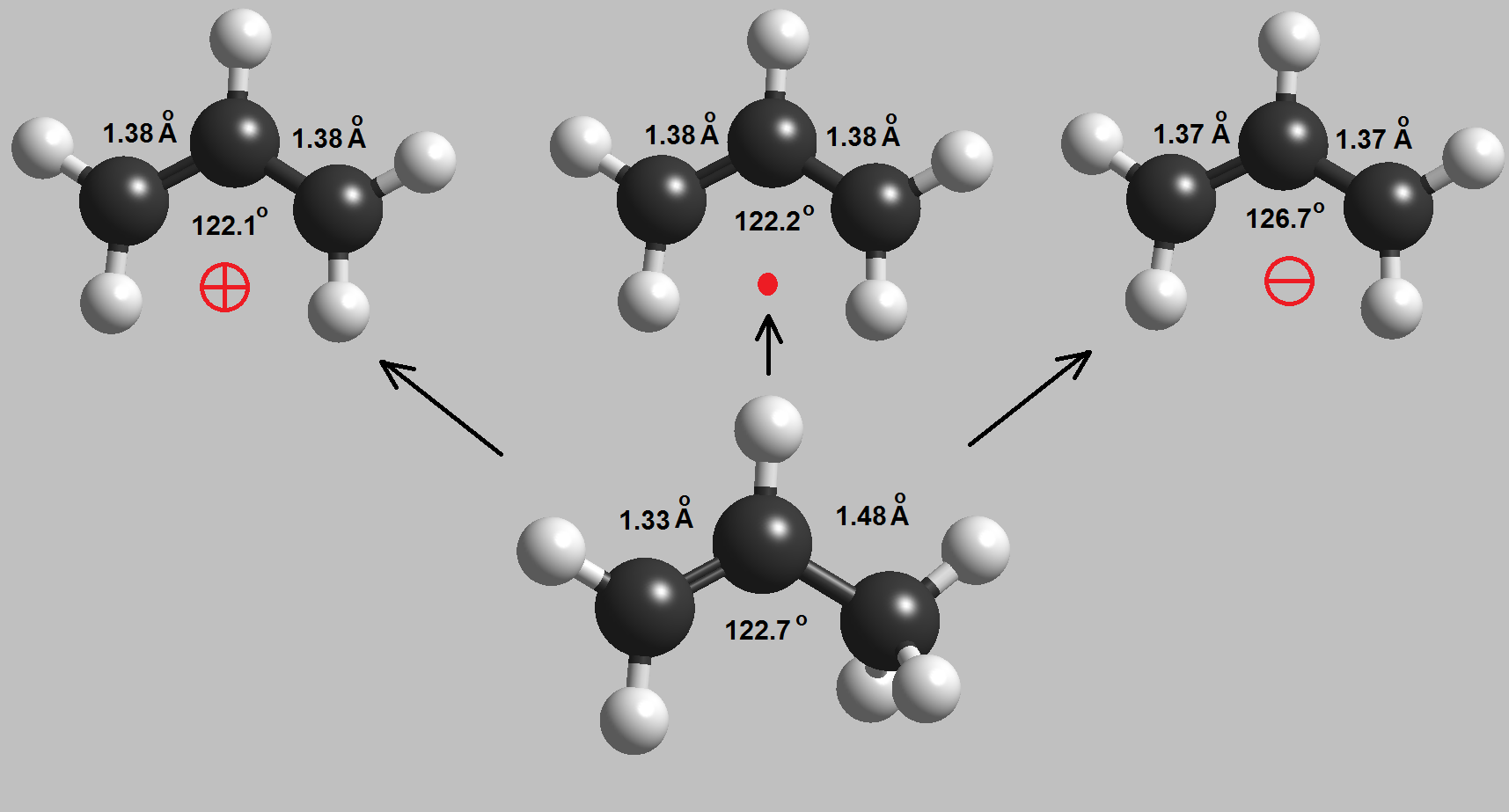
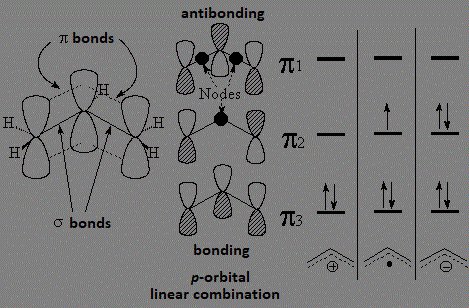
These resonant structures are indicating that the double C=C bond delocalizes the created species. A simple molecular orbital diagram enlightens us:
The three carbons of a allylic radical, carbocation or carbanion possess sp2 hybridization. The "p" orbitals on each of the three carbon overlap giving rise to three molecular orbitals where the charge or radical are delocalized.
This couldn' be possible hadn't the doble C=C bond been present.